Additional studies are needed to investigate this method. Several lines of evidence indicate that PP2, an inhibitor of non receptor tyrosine kinase c Src?a mediator in the EGFR signaling pathway?can abolish E2 induced Erk1/ 2 phosphorylation and, so, inhibit MCF 7 cell development. In our examine, GPR30 activation was inhibited by its particular antagonist G15, hence restraining proliferation of TAM R cells by initiating apoptosis beneath Tam interven tion. These outcomes are supported from the investigation of Ignatov et al, which indicated that GPR30 anti sense ol igonucleotides could eliminate GPR30 ligand mediated development stimulation of TAM R cells. During the in vivo research of your proliferative potential of GPR30, combin ation therapy of G15 plus Tam substantially reduced TAM R tumor dimension, whereas remedies with Tam or G15 alone did not.
GPR30 target remedy could boost apoptosis in TAM R xenografts, whereas apop tosis Checkpoint kinase inhibitor charges from Tam or G15 treatment don’t signifi cantly differ from that in the ethanol taken care of group. Synergistic interaction of GPR30 and the EGFR sig naling pathway enhances breast cancer proliferation, which allows tumor progression while in the presence of tamoxifen. Even though a number of endocrine resistant breast cancer designs are based upon inappropriate exercise with the EGFR signal ing pathway, the present model shows variable activation from the EGFR downstream cascade. Levels of phosphorylated Erk1/2 elevated transiently in our TAM R cells and in long run tamoxifen treated models reported by other folks. In contrast, sustained Erk1/2 phosphorylation was observed in long term estrogen deprived MCF 7 cells.
These variations might relate to methods that breast cancer cells adapt to various endo crine treatment options. selleck Although inappropriate activation with the EGFR signaling pathway is broadly accepted as being a key mechanism of tamoxifen resistance, the first aspect that transactivates EGFR is still disputed. Our research as a result aimed to demonstrate the function of GPR30 in the develop ment of tamoxifen resistance. In breast cancer MTs, GPR30 expression substantially enhanced relative to cor responding PTs and correlated with EGFR expression. Endocrine treatment method triggered greater ligand dependent activation in the EGFR downstream component Erk1/2, with consequential growth stimulation?which would lead breast cancer cells to build tamoxifen resistance. These phenomena were probably related to translocation of GPR30 for the cytomembrane and reduction of GPR30 induced cAMP production. As crosstalk concerning GPR30 along with the EGFR signaling pathway intensified, inhibited GPR30 exercise could encourage apoptosis 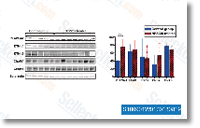 initi ation in drug resistant cells from the presence of tamoxifen.
initi ation in drug resistant cells from the presence of tamoxifen.
LPA Receptor Signaling
A protein that in humans is encoded by the LPAR1 gene.
